



The first corneal dystrophies were recorded 130 years ago by the German ophthalmologist Professor Arthur Groenouw. They are described as bilateral, symmetrical and slowly progressive inherited conditions unrelated to systemic or environmental factors. However, several corneal dystrophies violate this definition1.
Traditionally, these conditions are diagnosed based on clinical history and a stereotypical granular, macular, or lattice appearance. Although appearing as distinct entities, the growing genetic knowledge base for these diseases has revealed many to be closely related. Granular corneal dystrophy type 1 (GCD1) and type 2 (GCD2) and lattice corneal dystrophy (LCD1) have been found to all be caused by mutations of the same gene – transforming growth factor beta-induced (TGFBI)2. Genotyping has shown them to be closer to a disease spectrum than discrete entities3. Macular corneal dystrophy (MCD), however, has a unique genetic basis – a mutation of the carbohydrate sulfotransferase 6 (CHST6) gene.
This article focuses on GCD1, first described by Groenouw, and GCD2, known as Avellino dystrophy after the Italian province where it was discovered. These will be contrasted with LCD1 and MCD.
The TGFBI gene
The TGFBI protein (TGFBIp) is the second most common protein in the corneal stroma and is produced by epithelial cells and keratocytes4. Although TGFBIp exists throughout the body, pathologic forms only appear to accumulate in the cornea. This may be due to the high concentration of TGFBIp or specific factors present in the cornea’s extracellular matrix. Small changes in the amino acid composition of the pathologic proteins produce diverse clinical appearances, such as the refractile lines of LCD1 or the breadcrumb-like appearance of GCD1. Single amino acid changes can create misfolded or unfolded proteins which may form insoluble aggregates or altered breakdown products with the specific amino acid substitution, leading to the characteristic localisation of deposits found in the various TGFBI dystrophies1. Cultured corneal fibroblasts in GCD2 also show significant changes in gene expression. Consequent changes in the extracellular matrix may also contribute to a specific deposition pattern4.
GCD1
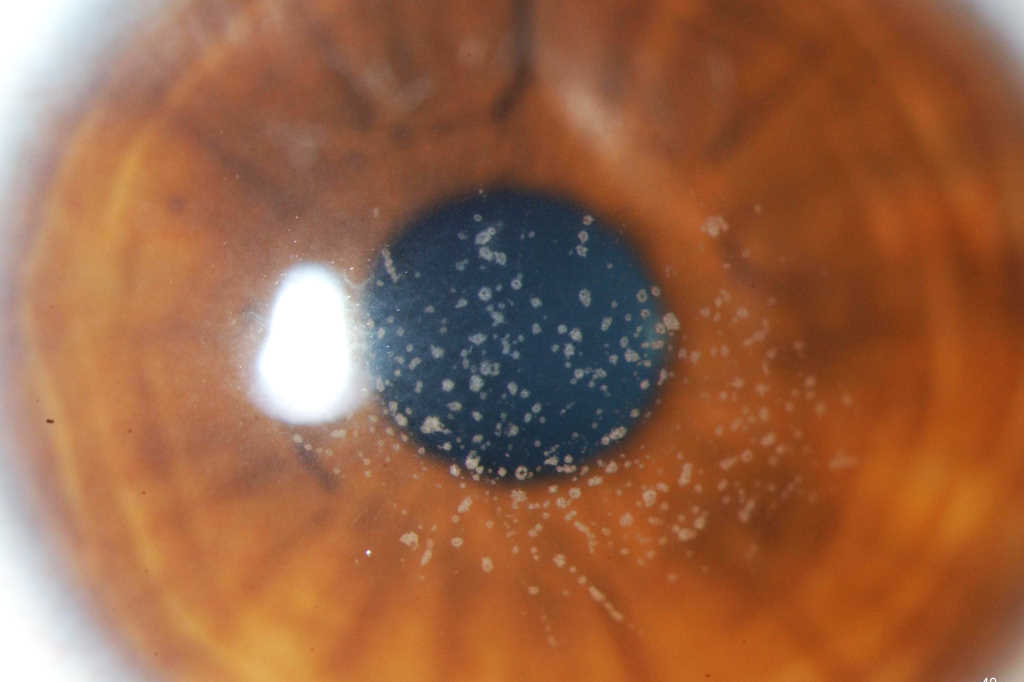
GCD1 is characterised by bilateral deposits in the anterior central stroma appearing as small, discrete, well demarcated, greyish-white opacities, typically appearing during the first or second decade of life. The deposits may first appear as fine dots which later take on a crumb-, ring-, or drop-shaped appearance. These lesions progressively increase in size, number, and may coalesce. Further advancement sees a ground-glass appearance of the cornea develop between lesions. In the early stages, patients normally suffer no discomfort or visual changes, although a subset does suffer erosive episodes. By the fourth decade, visual impairment can begin, typically secondary to opacification of the intervening stroma (Fig 1).
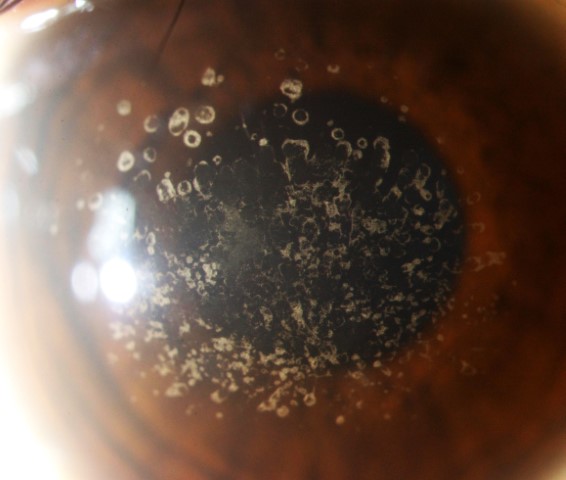
Fig 1a. Central crumb-like stromal deposits with clear intervening stroma
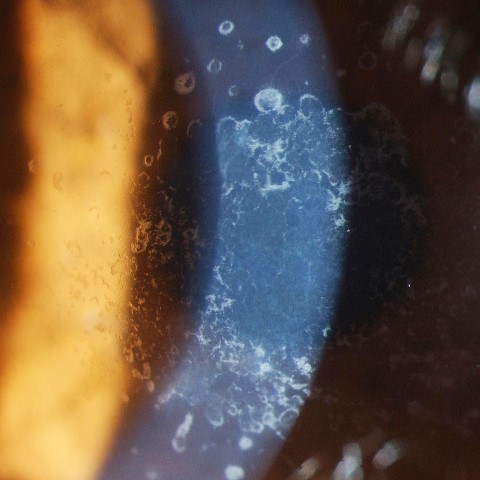
Fig 1b. Confluent breadcrumb granular opacities under direct and retro-illumination
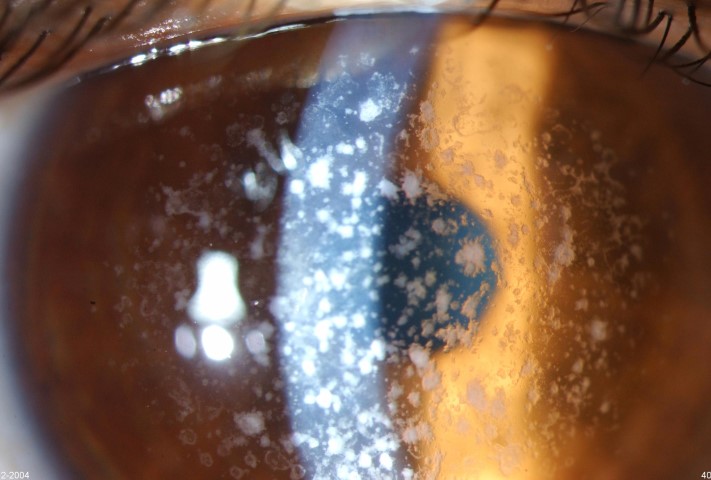
Fig 1c. GCD1 in the same family, with large peripheral-sparing grey-white opacities in the father
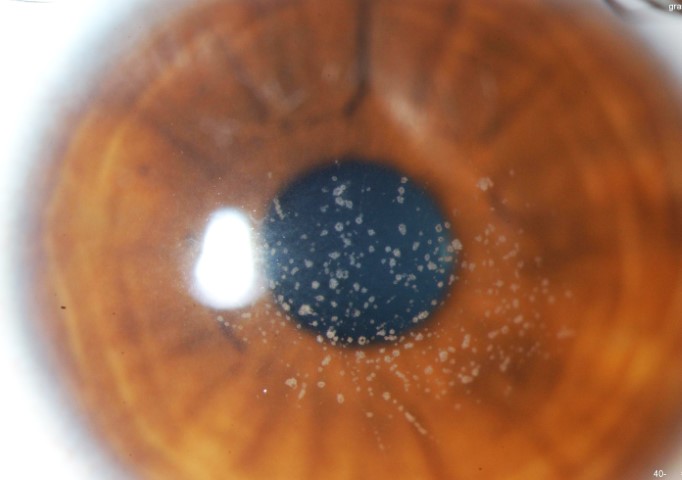
Fig 1d. GCD1 in the same family, with a much finer, granular appearance in the daughter.
Credit: Prof McGhee
GCD2
The stromal lesions in GCD2 are described as stellate and dendritic. They can, however, potentially look like those seen in GCD1. In addition, GCD2 is characterised by lattice lesions of the mid to posterior stroma and an anterior stromal haze. If lattice lesions appear, they always follow the presence of granular lesions. The stromal haze appears later in the course of the disease, with both these changes becoming more prominent with age. Recurrent corneal erosions occur frequently in these patients, so presentations of pain, photophobia and foreign-body sensation are much more common in GCD2 than GCD1. In both conditions, homozygotes for the affected gene suffer a more aggressive course.
Histopathologically, both present with rod-shaped or trapezoidal hyaline deposits beneath the epithelium, with GCD2 additionally demonstrating amyloid, which is related to the lattice lesions (Fig 2).
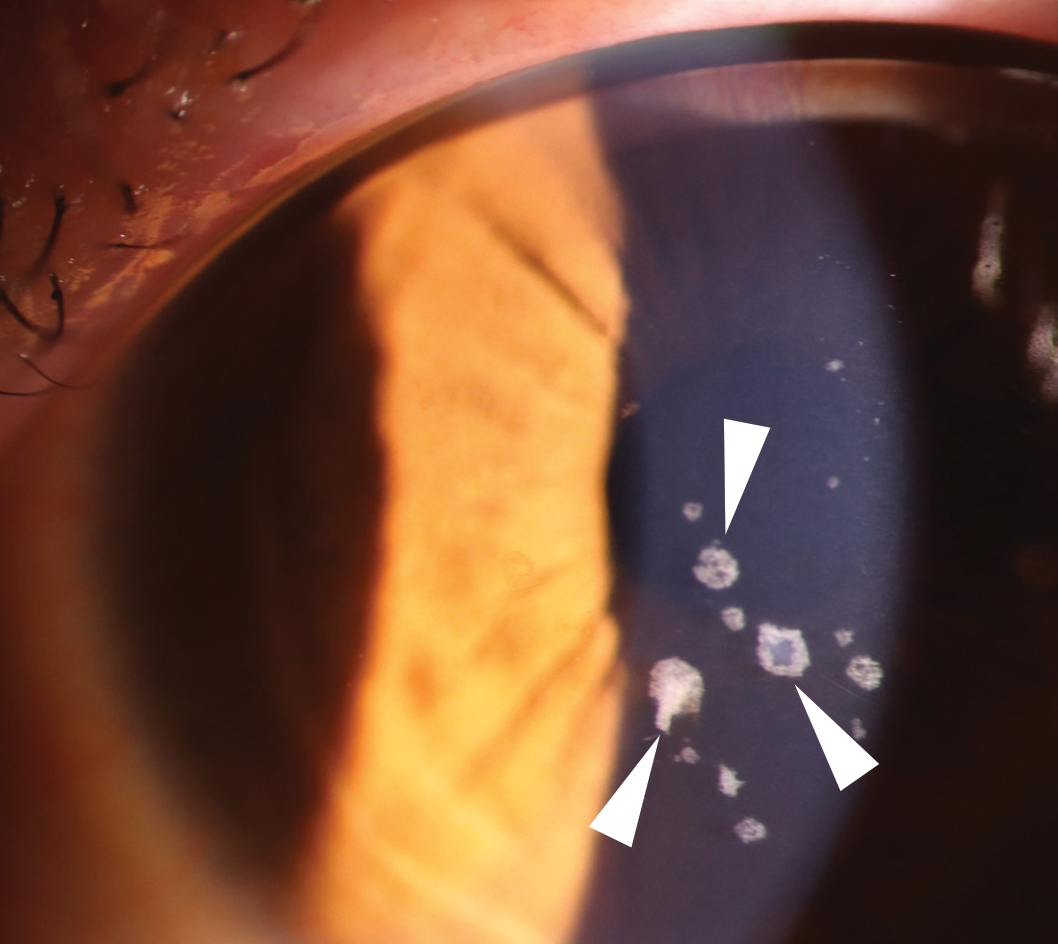
Fig 2a. GCD2 showing discrete granular opacities (arrowheads) in the corneal stroma centrally.

Fig 2b. Manifestation of GCD2 in the corneal interface postLASIK surgery, the eye only exhibited a few small grey lesions prior to surgery.
LCD1
LCD1 presents with branching lattice figures in the stroma with subepithelial opacities and an anterior stromal haze. This condition presents with considerable corneal variation, especially among the variant subtypes. This can make differentiation difficult, particularly with GCD2, which can also present with lattice lesions. Systemic disease can also occasionally present this way, such as in amyloidosis5. (Fig 3)
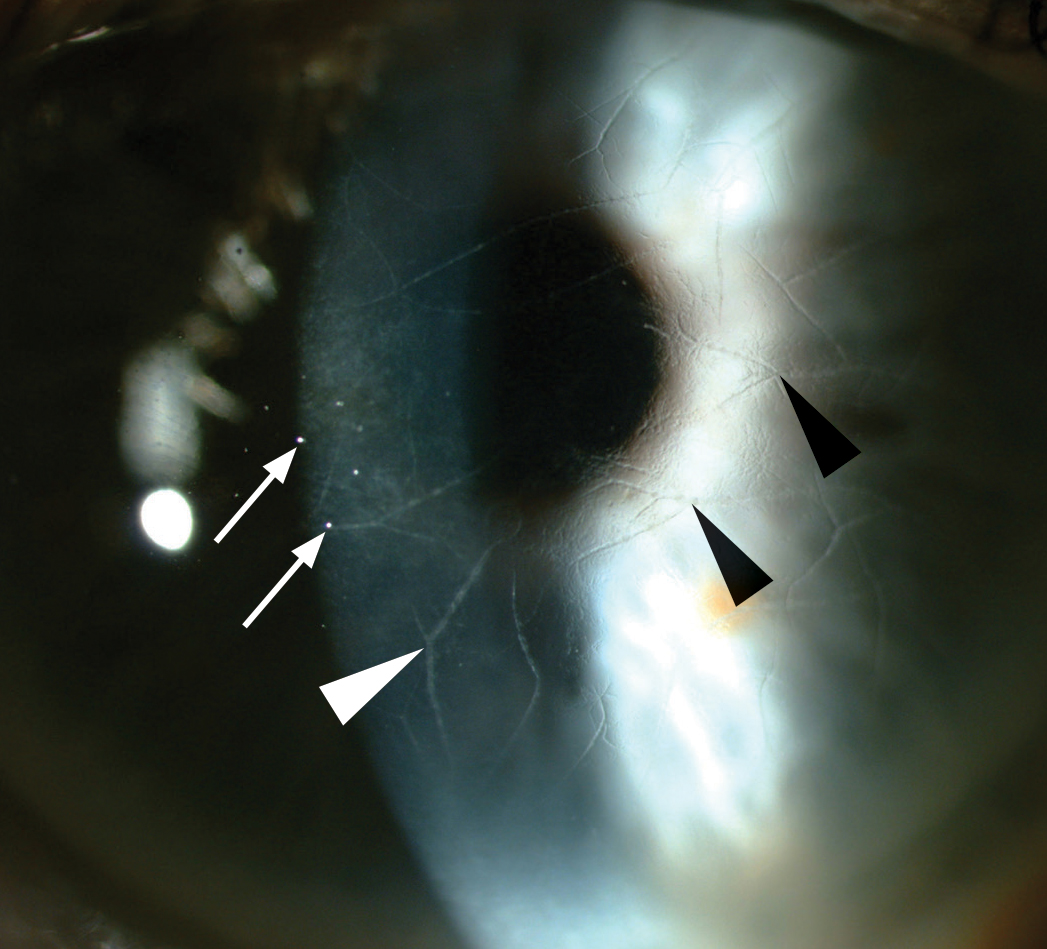
Fig 3. Refractile, branching lines (arrowheads) in the anterior corneal stroma with subepithelial ovoid white dots (arrows) and anterior stromal haze.
MCD
In its early stages, MCD can present similarly to granular dystrophy. However, it will often involve peripheral and deep layers of the stroma, which are typically spared with granular corneal dystrophies. An early stromal haze, thinning of the cornea and autosomal recessive family history supports this diagnosis (Fig 4). Table 1 highlights the differentiating features between these four granular dystrophies.
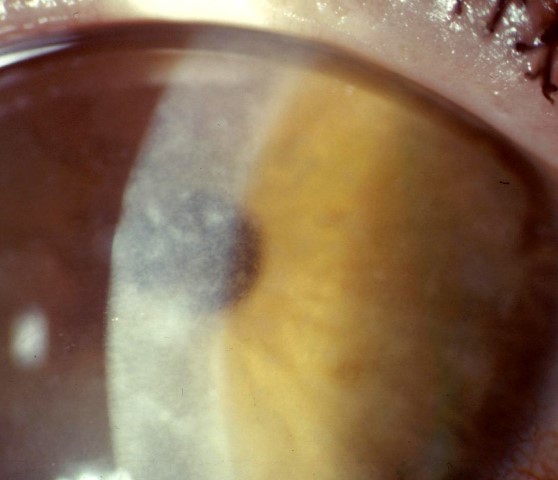
Fig 4a. MCD with extensive grey-white opacities throughout all corneal stromal levels
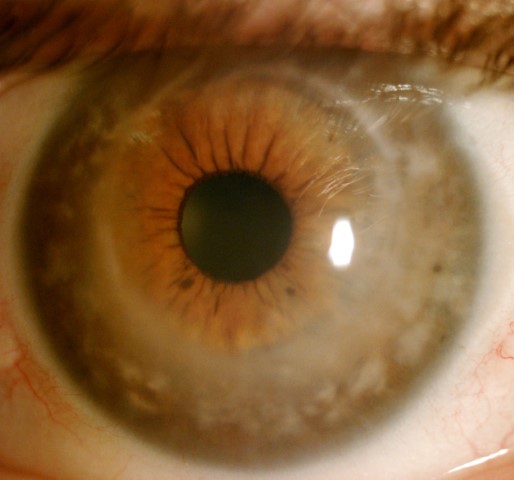
Fig 4b. MCD post corneal transplantation with a clear central cornea and stromal deposits in the periphery. Credit: All images, Prof McGhee
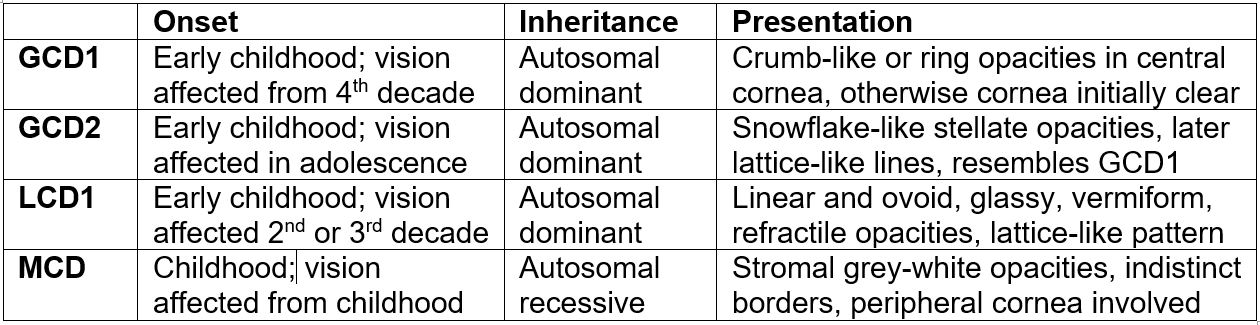
Table 1. Differentiating features between granular corneal dystrophy 1 and 2, lattice corneal dystrophy 1 and macular corneal dystrophy.
Conclusion
The corneal dystrophies discussed demonstrate a range of phenotypic appearances. While they are caused by a mutation in the same protein as LCD1, the specific amino acid substitution leads to a distinct clinical presentations. Conversely, MCD can appear similar to the granular dystrophies in the early stages, despite having an unrelated genetic basis. Phenotypic variation can be seen even among family members with the same dystrophy, so if these inherited conditions are suspected, it is important to examine relatives in attendance. Where the corneal appearance is equivocal, the pattern of inheritance, peripheral involvement and onset of visual loss can help clinically differentiate.
References

Scott Wilmshurst is a final-year medical student at the University of Auckland (UoA). He recently completed an elective in UoA and Auckland District Health Board ophthalmology services.
Dr Aaron Ong is a clinical research fellow in the Department of Ophthalmology, UoA.