




Functional evaluation in inherited retinal disease
Varela MD et al
Br J Ophthalmol Epub ahead of print: (25 Nov 2021)
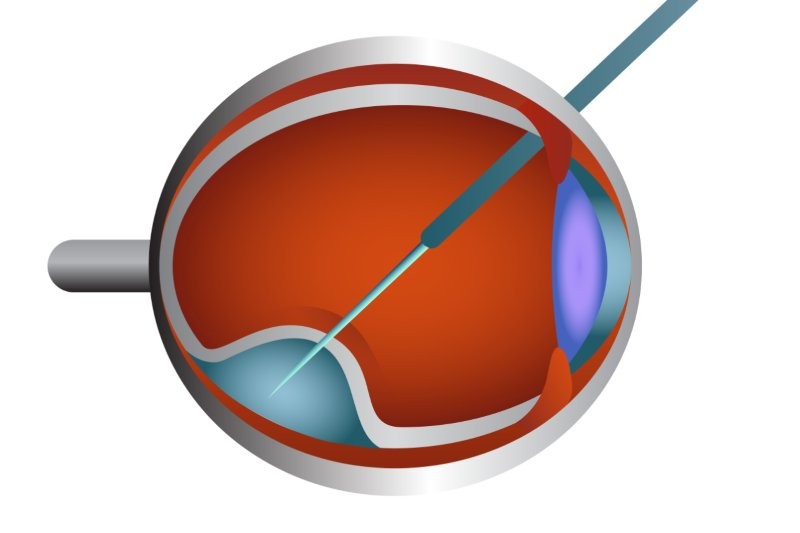
Review: A summary of the tools to document functional change in inherited retinal diseases (IRD), which are increasingly being used as outcomes in clinical trials for gene-targeted treatments. The rationale is that structural changes and optical coherence tomography (OCT), for example, are neither meaningful to the patient nor indicative of disease progression and treatment response.
Visual acuity (VA) remains a crucial parameter in the evaluation of function and integrity of the visual system; logMAR the most reliable using the Early Treatment of Diabetic Retinopathy Study (ETDRS) optotype, as well as low luminance VA using a 2.0 log unit neutral density filter, particularly in rod-cone dystrophies (RCD). Contrast sensitivity impacts reading speed and although there are limitations with the Pelli-Robson charts, it remains the gold standard. Colour vision is best measured with the Hardy-Rand-Rittler test, although computer-based systems such as the Cambridge colour test are frequently used in research.
Visual fields have moved away from kinetic perimetry, with the Octopus 900 semi-automated static perimetry the most robust to evaluate retinal sensitivity and VF integrity, and the Maia microperimeter important for macular function. Electrophysiology remains a crucial component, with the International Society for the Clinical Electrophysiology of Vision (ISCEV) standards, in addition to full-field light sensitivity threshold (FST). Patient-reported outcomes have increasing importance in clinical trial results, with the National Eye Institute (NEI) visual function questionnaire well validated.
Comment: With at least 50 clinical trials for IRD underway, establishing a natural history for individuals is crucial. The measures summarised in this paper not only aid diagnosis of the particular disease phenotype, but also emphasise the importance of regular annual review of IRD patients to establish their eligibility for clinical trials.
Juvenile-onset open-angle glaucoma – a clinical and genetic update
Selvan H et al
Survey of Ophthalmology (2021)
Review: The terminology, epidemiology and genetics and treatment of juvenile-onset open-angle glaucoma (JOAG), defined as onset from 4-40 years of age.
JOAG occurs more frequently in Africans and Indians; one study in Africa showed 16% of glaucoma cases were between 20 and 40 years of age, with a mean age at diagnosis of 16 years. Men are typically affected more and the disease can be unilateral. Intraocular pressure (IOP) is often higher at diagnosis – 30-40mmHg with wider diurnal variations of up to 22mmHg. Despite the nomenclature, angle abnormalities may be present in 60%, with a high iris insertion, with or without prominent iris processes, the most common manifestation. Disc haemorrhages and beta zone peripapillary atrophy are less common, but myopia is present in 42-72% with 30% over 6D.
Useful investigations include OCT angiography, demonstrating a strong positive correlation between retinal nerve fibre layer (RNFL) thickness and vascular density (VD) of disc and peripapillary region in JOAG. Usually presenting at moderate-advanced stages, VF at diagnosis shows baseline mean deviation of < -8.5dB.
MYOC, CYP1B1 and less commonly autosomal recessive LTBP2 have been associated with high-pressure JOAG. Optineurin (OPTN) and TANK-binding kinase 1 (TBK1) have been associated with juvenile normal tension glaucoma (JNTG).
Gene therapies are promising for JOAG caused by MYOC mutations and include trimethylamine n-oxide (TMAO) and sodium 4-phenylbutyrate, which rescues cells from apoptosis by alleviating endoplasmic reticulum (ER) stress. Genome editing using the CRISPR/Cas9 system reduced the expression of mutant MYOC and ER stress, resulting in lowered IOP and prevention of glaucoma development in a mouse model.
Comment: JOAG is not as infrequent as thought, particularly in certain ethnic groups. Because of the young age of onset, the projected disability from this disease is higher. Early diagnosis with knowledge of differences between JOAG and POAG, risk to other family members and optimal management, which may include gene-targeted therapies, can minimise the glaucomatous visual impairment in an age group at the peak of their productivity to society and their families.
Genetics of diabetic retinopathy
Bhatwadekar AD et al
Genes 2021, 12, 1200.
Review: Traditional risk factors, such as glycaemic control and duration of diabetes, are unable to explain why some individuals remain protected while others progress to a more severe form of the disease. Differences are also observed in diabetic retinopathy (DR) heritability as well as the response to anti-vascular endothelial growth factor (anti-VEGF) treatment. This review examines the genetic contribution to disease development.
Evidence for a genetic basis for DR was surmised due to higher prevalence in different racial groups (non-Hispanic Blacks, Mexicans and Native Americans), along with high heritability in family and twin studies. Candidate gene, linkage studies and genome-wide association (GWA) studies have identified associations with genes predisposing individuals to proliferative DR. Candidate gene studies suggest putative roles for AKR1B1, VEGFA, AGER, EPO and NOS3; however, some failed to replicate in populations with different ethnicities, again highlighting the role of genetics.
GWA studies have demonstrated that variation near growth factor receptor bound-2 (GRB2) is associated with sight-threatening DR. GRB2 is observed to be expressed by all human retina layers, including blood vessels, and an animal model showed that GRB2 is upregulated in DR. GRB2 binds phosphorylated insulin receptor substrate 1 and activates the mitogen-activated protein kinase (MAPK) pathway via Ras in response to insulin. GRB2 is also involved in VEGF signalling.
Another intergenic locus, rs476141, is situated between AKT serine threonine kinase 3 (AKT3) and zinc finger protein 238 (ZNF238). These genes play an essential role in cell survival, insulin signalling, and angiogenesis. A similar finding for the role of insulin signalling was reported in a GWA study performed in a Mexican population, where CAMK4 (calcium/calmodulin-dependent protein kinase IV) was found to be highly associated with DR, along with FMN1 (formin-1). CAMK4 increases transcriptional activity required for activating transcription factor-2 (ATF2) induced insulin gene expression.
Comment: A large number of genes are associated with the development of DR or PDR. Just as with macular degeneration and glaucoma, there is much work to be done, but the combination of different risk alleles (a polygenic risk score) may be able to give a more informed risk assessment for a given patient, as well as identifying therapeutic pathways for intervention, tailored to an individual’s genetic profile (precision medicine).

Dr Andrea Vincent is a clinician scientist and New Zealand’s first subspeciality ocular genetics ophthalmologist. She is an associate professor at the University of Auckland, undertaking research in genetic eye diseases, and established the Database of Inherited Retinal Disease of New Zealand. She also practices at the Eye Department of Greenlane Hospital and privately at Retina Specialists, Parnell.