



In December 2021, we described normal variations in the iris and iris changes in certain genetic conditions, systemic diseases, and malignant tumours. In the second part of this mini-series, we explore eye conditions resulting in iris transillumination defects and rubeosis.
Transillumination defects
Iris transillumination defects (ITD) can be elicited using the retro illumination technique which reflects light off the retina, during routine slit lamp examination1. It’s best conducted in a dim room where the patient’s pupils are mid-dilated, using a slit beam no higher than the diameter of the pupil and angled directly in line with the pupil1. The focus of the microscope is then brought to the iris at a magnification of 10 or 16x and any ITD or atrophy will appear as a red reflex (Fig 1A and B). ITD can be pathognomonic in several genetic conditions or can be a sequela following infections and trauma2. Commonly confused non-traumatic, progressive ITDs occur in iridocorneal endothelial (ICE) syndrome, Axenfeld-Rieger syndrome (ARS) and iridoschisis.
Genetic conditions causing ITD
Albinism is a disease characterised by little or no melanin in the tissues and demonstrates a distinctive ocular appearance. Albinism can be grouped into oculocutaneous albinism, which has an autosomal recessive inheritance, and ocular albinism, which has an X-linked inheritance pattern; both conditions affect the iris. The iris in albinism contains very little to no pigment to stop light reflecting from the retina and as such, diffuse ITD can be seen (Fig 1C)2. Aniridia is a rare, predominantly autosomal dominant, genetic disorder with variable degrees of ocular hypoplasia and/or iris absence. Despite the name, patients can have partial or complete absence of the iris (Fig 1D), alongside other ocular abnormalities such as cataract, glaucoma, corneal opacification and limbal stem cell deficiency. Another rare autosomal dominant disorder is ARS, a condition characterised by abnormal anterior segment development and systemic abnormalities such as mild craniofacial dysmorphism, dental abnormalities and redundant umbilical skin. Ocular examination of a patient with ARS often reveals ITD secondary to iris hypoplasia, corectopia and polycoria (Fig 1A and B). Other conditions causing ITD include anterior megalophthalmos, a spectrum of megalocornea involving other anterior segment abnormalities such as the iris (Fig 1E)3 and iridoschisis, a rare condition in which the anterior and posterior iris stroma and muscle layers are separated (Fig 1F). These two unique and rare conditions often present bilaterally.
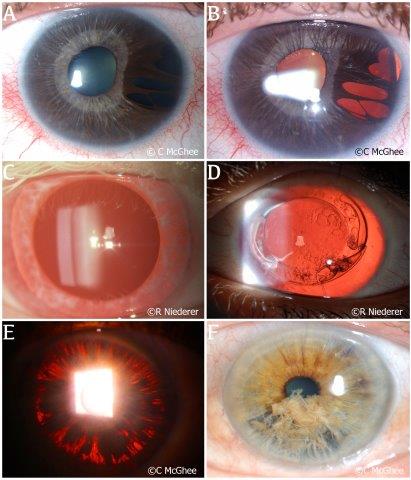
Fig 1. Genetic conditions causing ITD. (A) The iris of a patient with Axenfeld-Rieger syndrome in diffuse light. (B) The same iris examined showing the extent of multifocal iris atrophy under retro illumination. (C) Diffuse ITD in a patient with albinism. Note the fair depigmented eyelashes in this patient. (D) Cataract removal and intraocular lens implantation in a patient with complete aniridia. (E) Pigment dispersion on the iris surface due to chafing of the iris against the lens in an eye with anterior megalophthalmos. (F) A patient with iridoschisis demonstrating a ‘shredded’ appearance of the iris in diffuse light
Infections
Viral anterior uveitis – most commonly caused by herpes simplex virus (HSV) and varicella-zoster virus (VZV); less frequently cytomegalovirus, and rubella virus – can be complicated by atrophic iris changes resulting in ITD4. Herpes zoster ophthalmicus (HZO) describes the reactivation of VZV in the first division of the fifth cranial nerve (CN V1), which typically occurs in patients between the sixth to seventh decade of life, when VZV-specific immunity wanes4. In contrast, HSV uveitis, most commonly from HSV type-1, predominantly occurs in younger patients in the fourth to fifth decade of life and is caused by the reactivation of the HSV virus from within the trigeminal ganglion. Up to 60% of patients with HZO experience ocular involvement and these patients generally experience more inflammation than those with HSV-related uveitis. Segmental (Fig 2A) or patchy (Fig 2B) atrophy causing ITD can develop in as many as 88% of HZO patients and 48% of patients with HSV4.
Trauma
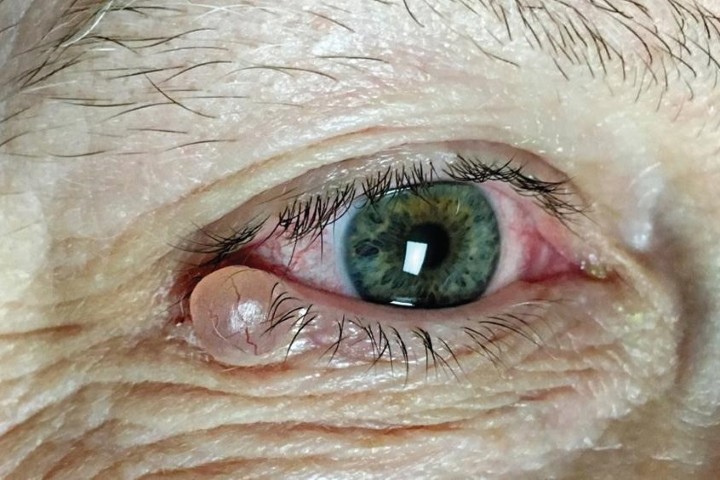
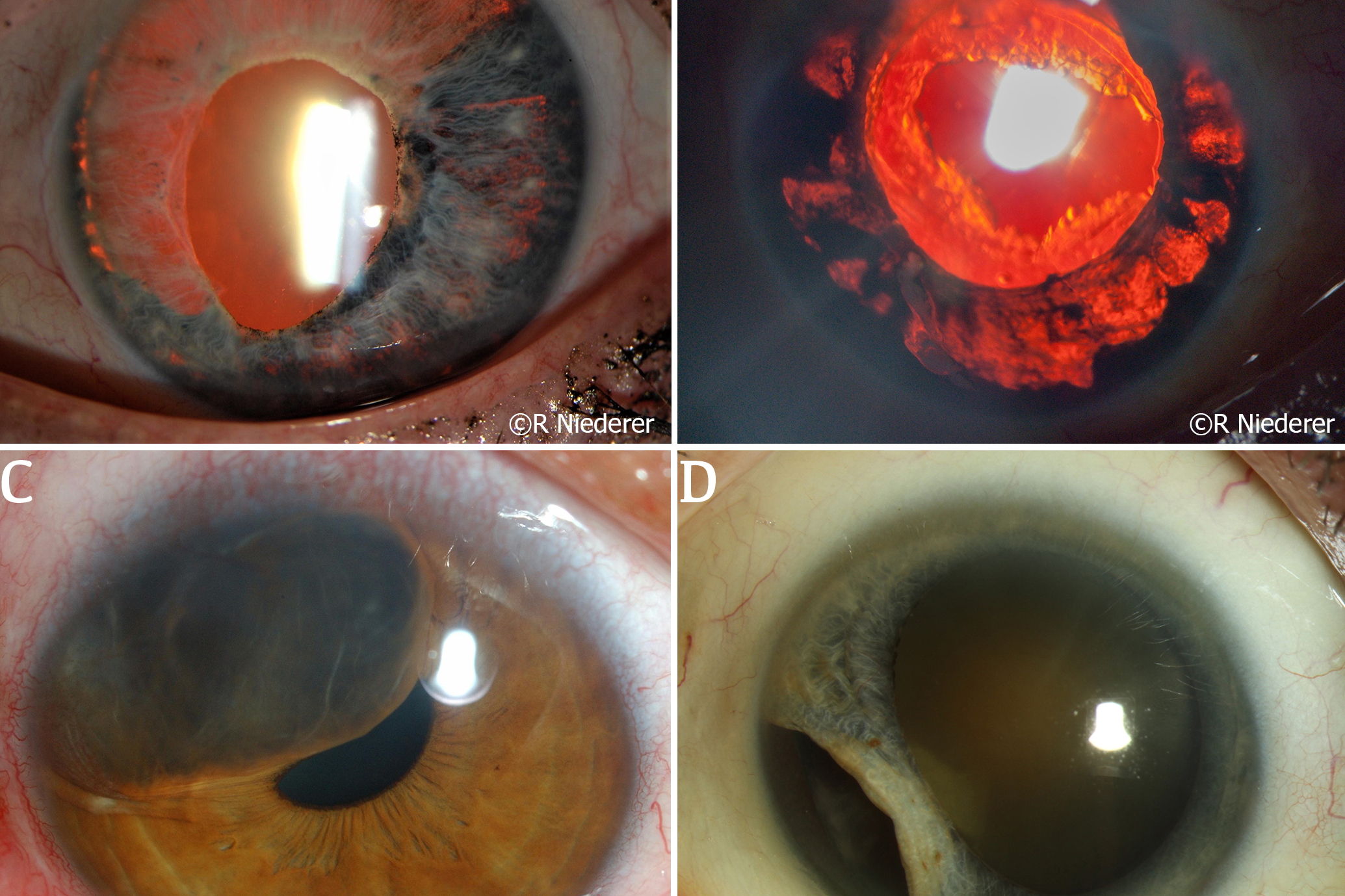
Traumatic ITD may result from penetrating eye injuries, blunt trauma, or iatrogenic causes intraoperatively. In an acute setting, a history of any acute trauma resulting in an ITD should prompt a clinician to carefully assess the status of the structures posterior to the iris, specifically looking for signs of a traumatic cataract, commotio retinae, or presence of any intraocular foreign body. A missed intraocular foreign body may result in infection (endophthalmitis), inflammation (uveitis) or traumatic cysts (Fig 2C), requiring further intervention. Trauma from the above mechanisms can also result in iris avulsion from the iris root, leading to iridodialysis (Fig 2D), which can result in cosmetic and functional issues such as glare and monocular diplopia5. Regardless of the mechanism, any damage to the iris may have consequent damage to the drainage angle and increases the patient’s risk of developing glaucoma.
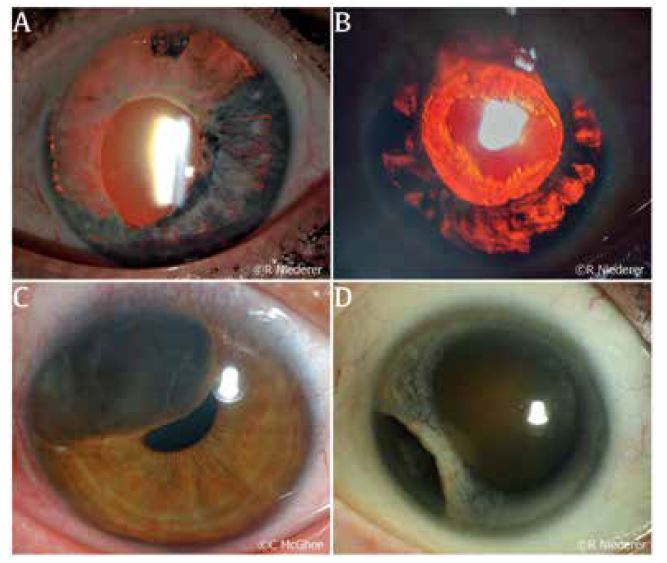
Fig 2. (A) A patient with HSV anterior uveitis and resultant large segmental atrophy seen with retro illumination. (B) A patient with ocular-involving herpes zoster ophthalmicus; severe anterior uveitis resulted in patchy iris atrophy seen as ITDs. (C) Traumatic cyst displacing iris structures towards the pupil. (D) Iridodialysis following blunt trauma to the eye of a patient
Other differentials causing ITD
Other differentials to consider, not discussed in detail above, include pigment dispersion syndrome, pseudoexfoliation syndrome and Fuchs’ heterochromic iridocyclitis. These conditions have an unknown aetiology and are likely to be multifactorial, with the most common complication being glaucoma from angle involvement. In the last two decades, a new disease entity known as bilateral acute iris transillumination (BAIT) syndrome has been recognised. The main complaint from patients with BAIT is strong photophobia caused by spectacular transillumination of the iris and observed to occur in patients following upper respiratory tract infections, and/or use of oral moxifloxacin prior to development of BAIT6.
Rubeosis iridis
Rubeosis iridis is the condition of iris neovascularisation, first described by Coats in 1906. When uncontrolled, rubeosis iridis can lead to neovascular glaucoma, a disease entity that progresses quickly, is sight threatening and has poor visual prognosis (Fig 3A)7,8. Little is published on the aetiology of rubeosis iridis8 and, in existing studies, its pathogenesis is thought to be predominantly driven by retinal ischaemia. The ischaemic posterior segment has been shown to trigger a cascade of events leading to an increase in pro-angiogenic factors such as vascular endothelial growth factors (VEGF) and interleukin-6, alongside a decrease in anti-angiogenic factors, such as pigment-epithelium-derived factor7. The imbalance results in proliferation of new vessels in the anterior segment resulting in the clinical finding of rubeosis iridis. If left untreated, the condition can progress rapidly to neovascular glaucoma from as early as two weeks to 12 months, depending on the aetiology.
The vast majority of rubeosis iridis cases are caused by central/branch retinal vein occlusion (RVO), proliferative diabetic retinopathy (Fig 3B), ocular ischaemic syndrome and central retinal artery occlusion (CRAO) (Fig 3C)7,8. Less common but significant causes of rubeosis iridis include ocular tumours (uveal melanoma, retinoblastoma) (Fig 3D), systemic diseases (systemic lupus erythematosus, juvenile xanthogranuloma) and other causes such as uveitis, radiation treatment and chronic retinal detachment7,9. The new vessels formed in this condition do not follow an organised growth pattern and are more prone to leakage or damage due to the lack of endothelial integrity. As a result, patients can develop acute spontaneous hyphaema (Fig 3C) and this in itself can lead to angle recession in an eye where the trabecular meshwork is already compromised by neovascularisation.
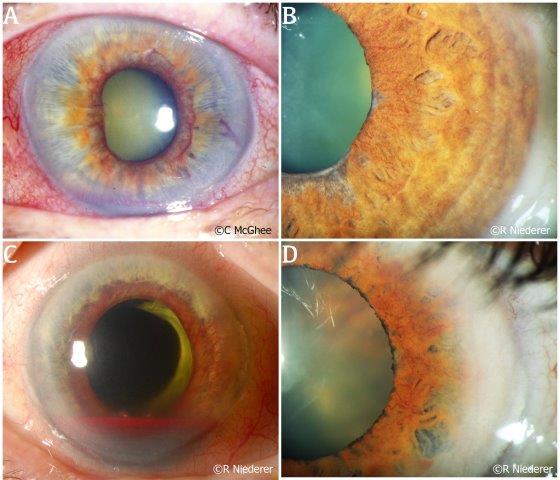
Fig. 3 (A) Rubeosis iridis in a patient who has progressed to neovascular glaucoma. (B) Subtle but diffuse neovascularisation at the pupillary margin of the iris observed in a patient with advancing diabetic retinopathy. (C) Rubeosis iridis in a patient with central retinal artery occlusion. This patient also experienced a hyphaema secondary to bleeding from the neovascularisation. (D) Subtle rubeosis iridis in a patient with choroidal melanoma
Early recognition of rubeosis iridis is key in determining the trajectory of the patient’s visual prognosis. The presence of any new vessels on the iris should prompt immediate treatment to reduce r halt the ischaemic process in the retina. This can be achieved by pan-retinal photocoagulation of the diseased retina to diminish the burden of ischaemia or by medical treatment with intravitreal anti-VEGF to reduce the proliferation of new vessels.
Conclusion
The iris is truly a window to the health of the human body. Many genetic or acquired conditions may manifest first in the eye and be visualised using the slit lamp. Additional slit-lamp techniques, such as the retroillumination technique described in this series, when employed routinely, can further elicit subtle intraocular findings, leading the clinician to the diagnosis. Early recognition of iris transillumination defects and presence of rubeosis iridis can change the visual prognosis of a patient by allowing timely and necessary interventions.
References
We would like to acknowledge Professor Charles McGhee, head of the Department of Ophthalmology at the University of Auckland (UoA), and Dr Rachael Niederer for their generous contributions of illustrative case images to these articles.

Dr Joevy Lim is a clinical research fellow and PhD candidate studying the status of ocular melanoma in NZ. She was awarded an HRC Clinical Research Fellowship Training grant last year for her ongoing research and has special interests in ocular oncology and anterior segment disorders.

Dr Jie Zhang is a senior HRC research fellow and scientist in the Dept of Ophthalmology, UOA. She has specific research interests in both laboratory and clinical aspects of the cornea and anterior segment.

Dr Rachael Niederer is a senior lecturer in the Dept of Ophthalmology, UOA and a consultant ophthalmologist with a special interest in clinical and research aspects of uveitic eye conditions.